Эктодермальная дисплазия
OMIM 305100
Наша команда профессионалов ответит на ваши вопросы
Ангидротическая эктодермальная дисплазия (синдром Криста-Сименса-Турена) — наследственное заболевание, характеризующееся врожденной гипоплазией потовых желез, волосяных фолликулов, желез слизистых оболочек и аплазией большинства зубных зачатков, дисморфогенезом мягких тканей полости рта. Дисморфогенез мягких тканей лица при этом заболевании включает изменения языка. Он увеличен, складчатый, сухой, нередко на спинке языка имеется трудно снимаемый налет. Сосочки на кончике языка сглажены. Дно полости рта расположено высоко, подъязычные железы небольшого размера. Слюна выделяется скудно, из-за чего больные испытывают постоянно сухость во рту, всегда при еде запивают пищу водой. Также у пациентов наблюдается непереносимость высокой температуры из-за недоразвития потовых желез, кроме того, в зеве, трахее, гортани и бронхах отсутствуют слизистые железы, из-за чего больные восприимчивы к бактериальным инфекциям. Частота встречаемости эктодермальной ангидротической дисплазии составляет 1 на 100 000 новорожденных.
Наиболее распространенной причиной заболевания является повреждение гена, кодирующего белок эктодисплазин-А (EDA). Ангидротическая эктодермальная дисплазия наследуется по Х-сцепленому рецессивному типу с более тяжёлым течением у мужчин. Ген эктодисплазина-А расположен на длинном плече Х хромосомы (Хq12-q13.1). В настоящее время описано более 200 мутаций в этом гене, включая крупные делеции. В Центре Молекулярной Генетике проводится прямая ДНК-диагностика эктодермальной ангидротической дисплазии, заключающаяся в поиске мутаций в гене EDA методом прямого автоматического секвенирования.
Однако в некоторых случаях данного метода бывает недостаточно для выявления мутации или для определения носительства мутантного гена у женщин. Так, например, у матерей, для сыновей которых была выявлена делеция всего гена EDA или его части, невозможно с помощью секвенирования определить носительство мутации вследствие наличия второй интактной Х-хромосомы. В этом случае показано определения числа копий гена EDA методом количественной мультиплексной лигазной реакции. Для анализа необходимо предоставить ТОЛЬКО свежую жидкую несвернувшуюся нелизированную кровь в пробирке с ЭДТА.
В Центре Молекулярной Генетике также проводится анализ числа копий гена EDA, а также генов EDAR (2q13) и EDARADD (1q43), мутации в которых приводят к эктодермальной ангидротической дисплазии с аутосомно-доминантным или рецессивным типами наследования.
На базе ФГБУ «ЦННИС и ЧЛХ», отделения челюстно-лицевой хирургии проводится зубочелюстная реабилитация пациентов с подтвержденным диагнозом: эктодермальная дисплазия. Реабилитация включает многоэтапное хирургическое лечение в объёме:
1)костная пластика (увеличение объёма костной ткани на верхней и нижней челюсти)
3)зубочелюстное протезирование (изготовление зубного протеза на верхнюю и нижнюю челюсть)
Для получения квоты на хирургическое лечение необходимо иметь молекулярно-генетическое подтверждение диагноза «эктодермальная ангидротическая дисплазия»
Контактный email для консультации по вопросам лечения:
ponomarev_100@list.ru (Пономарёв Артемий Эрнестович)
Медицинские интернет-конференции
Языки
Симптомокомплекс эктодермальной дисплазии в клинике стоматологии
Торгашина А.Г., Фирсова И.В.
Резюме
В данной статье на конкретном клиническом примере представлен симптомокомплекс эктодермальной дисплазии, стоматологическое лечение и способы профилактики при данной патологии.
Ключевые слова
Статья
Описаны около 175 клинически и генетически различных форм эктодермальной дисплазии (Фрейре-Майя и Пиньейру). Каждый вид эктодермальной диплазии обычно включает в себя различные комбинации симптомов, которые могут варьироваться от легких, до тяжелых, таких как отсутствие или уменьшение количества потовых желез, отсутствие или аномалия роста волос, отсутствие или порок развития некоторых или всех зубов. При эктодермальной дисплазии возможно ослабление или утрата слуха/зрения, отсутствие или порок развития некоторых пальцев рук или ног, расщелина губы и/или неба, нерегулярная пигментация кожи, чувствительность к свету, проблемы с органами дыхания, отсутствие развития молочных желез. Кроме того часты инфекции из-за иммунодефицита, а в некоторых случаях, из-за неспособности поврежденной кожи (трещины, эрозия) сопротивляться болезнетворным бактериям, и другие симптомы.
К наиболее часто встречающимся проявлениям эктодермальной дисплазии относятся гипогидротическая (dysplasia ectodermalis hydrotica) и ангидротическая (dysplasia ectodermalis anhydrotica) формы, а также ряд синдромов, в которых кожные дефекты ассоциированы с другими врожденными аномалиями (синдром Marchall, синдром Helweg-Larsen и др.).
Средние данные по распространенности эктодермальной дисплазии в мире варьируются от 1:10 000 до 1:100 000. Показатель гипогидротической эктодермальной дисплазии составляет 1:17000 новорожденных.
Ангидротическая эктодермальная дисплазия (синдром Криста-Сименса-Турена) впервые была описана в 1848 году J.Thuraine [8].
Характерные клинические признаки ангидротической эктодермальной дисплазии.
Низкий рост, старческий вид лица. Лицо имеет особенности: большой лоб с выступающими надбровными дугами и лобными буграми («олимпийский лоб»), широкие скуловые кости, запавшая переносица, маленький седловидный нос с гипоплазией крыльев, запавшие щеки, полные вывернутые губы, массивный подбородок, большие деформированные уши («уши сатира»).
Кожа сухая, истонченная (78%). Потоотделение снижено (85%) или реже отсутствует по всему кожному покрову, за исключением мест локализации апокриновых желез, где оно в большей или меньшей степени может сохраняться, непереносимость жары (50%) [9].
Недоразвитие секреторных желез приводит к возникновению конъюнктивитов (из-за уменьшения слезоотделения), ларингитов, фарингитов, ринитов (в том числе атрофического), стоматитов. Эти же железы недоразвиты в слизистой оболочке трахеобронхиального дерева, пищевода и двенадцатиперстной кишки, что клинически проявляется рецидивирующими легочными инфекциями, охриплостью и дисфагией [11].
Волосы редкие, тонкие, сухие, медленно растущие (89%); редкие брови (96%), отсутствие ресниц (100%). Скудное оволосение (62%) в подмышечных областях и на лобке. Пушковые волосы обычно сохраняются. Возможна тотальная аллопеция, ониходисплазия (39%) [9].
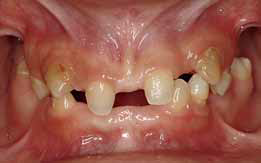
Проявления в полости рта: гиподонтия, анодонтия, микродонтия, или другие аномалии зубов (100%). Зубы поздно прорезываются, долго сохраняются на стадии молочных, могут быть не в полном количестве, или даже отсутствовать полностью, часто деформированы, с большими промежутками между ними. За счет деформации зубов кожа вокруг рта может иметь складки, напоминающие рубцы при врожденном сифилисе. Старческий вид пациентов обусловлен снижением нижней трети лица при полном отсутствии зубов и резкой выраженностью супраментальной складки [6].
У ряда больных наблюдается ихтиозиформное шелушение, фолликулярный гиперкератоз, ладонно-подошвенные кератодермии.
Реже отмечают стеноз слезной точки, дисплазию роговицы, катаракту, аномалию гонад и тугоухость по проводящему типу [7].
Из других состояний описана склонность к атипическим заболеваниям, экзематозным реакциям. Возможны атопический дерматит и астма, гипотиреоз, папулезные высыпания, обусловленные сально-железистой гиперплазией. У женщин заболевание протекает в смягченной форме в виде не резко выраженных зубных аномалий, очаговых расстройств потоотделения, слабого развития молочных желез [2].
Умственное развитие у большинства больных нормальное, иногда выявляются признаки снижения интеллекта.
Иллюстрацией к вышеизложенному, может служить описание пациента с ангидротической эктодермальной дисплазией, родители которого обратились на кафедру стоматологии детского возраста и ортодонтии СГМУ им. В.И.Разумовского с жалобами на полное отсутствие временных зубов у ребенка.
Пациент А., 2,1 года. При внешнем осмотре: лицо имеет старческий вид. Отмечается уменьшение нижней трети лица; уменьшение гнатической области лица, резко выраженная супраментальная складка, лоб с выступающими надбровными дугами и лобными буграми; ушные раковины слегка деформированные. Волосы светлые, короткие, тонкие, сухие, редкие. Брови и ресницы отсутствуют. Кожа сухая, бледная, с морщинками на верхних и нижних веках.
При осмотре полости рта отмечается полное отсутствие зубов. Слизистая оболочка полости рта сухая, бледная. Верхняя и нижняя челюсти значительно уменьшены с резко выраженным недоразвитием альвеолярных отростков. Соотношение беззубых альвеолярных отростков нормогнатическое (Рис.1,2).
Ребенок находится на диспансерном учете у эндокринолога.
При проведении рентгенологического обследования на ОПТГ обнаружены зачатки молочных и постоянных центральных резцов верхней челюсти: гиподонтия временных и постоянных зубов.
На основании полученных данных поставлен диагноз: эктодермальная ангидротическая дисплазия, множественная первичная адентия временных и постоянных зубов тяжелой степени, задержка прорезывания зубов 51, 61.
Лечение симптоматическое. Эктодермальная ангидротическая дисплазия, сопровождающаяся множественной или полной первичной адентией, является показанием раннего ортопедического лечения [5].
Данному пациенту рекомендовано изготовление полного съемного протеза на верхнюю и нижнюю челюсть, с последующей своевременной заменой в связи с ростом и развитием челюстно-лицевой области. В дальнейшем рекомендовано изготовление частично съемного протеза на верхнюю челюсть в случае прорезывания центральных резцов.
Ортопедическое лечение восстановит высоту нижней трети лица и как следствие улучшит внешний вид, нормализует функцию жевания и ускорит психологическую адаптацию пациента.
Рекомендовано орошение слизистой оболочки полости рта и носа. Использование искусственной слезной жидкости помогает предотвратить повреждения роговицы у больных с недостаточным слезоотделением. С косметической целью при алопеции может потребоваться ношение парика.
В профилактических целях в случае планирования последующей беременности в данной семье необходимо проведение медико-генетического консультирования, для определения степени риска рождения следующего ребенка с подобной патологией.
Эктодермальной дисплазией в семье болеют только мальчики. Девочки будут являться облигатными носителями и могут показать минимальные проявления.
Риск для сибсов зависит от генетического статуса родителей.
Мужчина с синдромом эктодермальной дисплазии будет передавать мутированный ген только своим дочерям. Женщина с синдромом эктодермальной дисплазии будет передавать ген, локализованный в Х хромосоме своим потомкам независимо от пола. Ее сыновья будут иметь 50%-й риск проявления синдрома с дальнейшей передачей гена. У ее дочерей есть 50% риск оказаться носителями патологического гена с минимальными проявлениями в фенотипе. Мутированный ген может проявиться в дальнейших поколениях у лиц мужского пола.
Таким образом, на конкретном клиническом примере мы рассмотрели проявления эктодермальной ангидротической дисплазии. Данная патология требует комплексного врачебного подхода. Стоматологическое лечение заключается в длительном наблюдении у ортодонта. Раннее зубочелюстное протезирование необходимо для нормализации жевательной функции, деятельности ЖКТ и улучшения эстетического вида пациента.
Литература
2. Смердина Ю.Г., Смердина Л.Н. Генезис и клиника эктодермальной дисплазии ангидротической (синдром Криста–Сименса–Турена) // Медицинские науки. 2008. №5. С.138-139.
3. Сивовол С.И. Симптомы, синдромы, эпонимные болезни челюстно-лицевой области, головы и шеи. М.: Триада-Х. 2002. С.93-94.
4. Миллет Д. Решение проблема в ортодонтии и детской стоматологии / Д.Миллет, Р.Уэлбери; пер. с англ. М.: МЕДпресс-информ. 2009. С.143.
5. Гепалова К.В., Чабан А.В. Этапы лечения аномалий количества и сроков прорезывания зубов при врожденных синдромов: ангидротической эктодермальной дисплазии ключично-черепном дизостозе Шейтхауэра-Мари-Сентона: клинический случай // Актуальные проблемы стоматологии детского возраста. Сборник научных статей I региональной научно-практической конференции по детской стоматологии. Хабаровск. 2011. С.40-42.
6. Коминек Я., Томан Я., Розковцова Е. Проявления дегенеративных заболеваний в ротовой полости // Детская стоматология. Прага. Государственное издательство медицинской литературы. 1968. С.417-418.
7. Eismann H., Knauer K., Kunzel W., Muller M., Muller W. Storungen der Dentition und Zahnenntwicklung // Kinderrstomatologie. Leipzig. 1988. P.136-139.
8. Zonana J., Elder M.E., Schneider L.C., Orlow S.J., Moss C., Golabi M., Shapira S.K., Farndon P.A., Wara D.W., Emmal S.A., Ferguson B.M. A Novel X-Linked Disorder of Immune Deficiency and Hypohidrotic Ectodermal Dysplasia Is Allelic to Incontinentia Pigmenti and Due to Mutations in IKK-gamma(NEMO) // Am. J. Hum. Genet. 2000. № 67(6). Р.55–62.
9. Lingling Ho, M.S. Williams, R.A. Spritz. A Gene for Autosomal Dominant Hypohidrotic Ectodermal Dysplasia (EDA3) Maps to Chromosome 2q11-q13 Am. J. Hum. Genet. 62:1102–1106, 1998.
10. Bala M., Pathak A. Ectodermal dysplasia with true anodontia // J. Oral Maxillofac. Pathol. 2011. № 15(2). Р. 44–46.

Ангидротическая эктодермальная дисплазия – это наследственное заболевание, проявляющееся генетическим нарушением развития производных эктодермы (кожи, желез внешней секреции, волос, зубов). Симптомами патологии являются аномалии развития или отсутствие зубов, сухая тонкая кожа, выраженная гипоплазия потовых и сальных желез, редкость волос или алопеция, иногда агенезия молочных желез. Диагностика производится врачом-генетиком на основании характерных внешних проявлений заболевания, а также генетических исследований и изучения наследственного анамнеза. Специфического лечения ангидротической эктодермальной дисплазии не существует.
МКБ-10

Общие сведения
Ангидротическая эктодермальная дисплазия (синдром Криста-Сименса-Турена, синдром Weech) является наследственным заболеванием, при котором происходит генетически обусловленное нарушение развития наружного зародышевого листка (эктодермы). В результате этого основные симптомы патологии затрагивают производные эктодермы – кожу, волосы, зубы, некоторые хрящи, потовые, сальные и молочные железы. Характерный для ангидротической эктодермальной дисплазии симптомомкомплекс независимо друг от друга описывали Дж. Турен в 1848 году, стоматолог Дж. Крист в 1913-м и дерматовенеролог Х. Сименс в 1929-м году.
Первоначально считалось, что наследование заболевания происходит исключительно по сцепленному с Х-хромосомой механизму, в настоящее время выявлены как аутосомно-рецессивные, так и аутосомно-доминантные формы дисплазии. Встречаемость заболевания – 1 случай на 5000-10000 тысяч новорожденных, с учетом того, что статистически чаще встречается сцепленное с полом наследование, половое распределение среди больных сильно смещено в сторону мужского пола.
Причины
Этиология ангидротической эктодермальной дисплазии заключается в наличии мутаций определенных генов. Различают формы синдрома Криста-Сименса-Турена с Х-сцепленным, аутосомно-доминантным и аутосомно-рецессивным наследованием:
Симптомы
Возникшая гипоплазия кожи и многих типов желез (потовых, слезных, молочных) ведет к каскаду разнообразных нарушений. Практически полное отсутствие потовых желез становится причиной легкого развития гипертермии, что особенно опасно в детском возрасте – именно из-за последствий перегрева в раннем детстве умирает почти треть больных ангидротической эктодермальной дисплазией. В результате пониженной активности слезных желез достаточно часто возникают конъюнктивиты, которые осложняются кератитом и катарактой.
Гипопластическая кожа довольно часто подвержена экземе, вторичным бактериальным и грибковым инфекциям. Нарушения развития эктодермы отражаются и на хрящевых и костных элементах – увеличивается размер лобной кости, на ней формируются заметные надбровные дуги. Переносица запавшая, крылья носа, как правило, недоразвиты, деформируются и ушные раковины.
Типичны аномалии зубов, которые приобретают коническую форму, часто бывают недоразвиты, возможно отсутствие одного или целой группы зубов; характерным признаком ангидротической эктодермальной дисплазии при этом является сохранение клыков. Из-за отсутствия или аномального положения зубов нередко развиваются дефекты речи.
Интеллектуальное развитие ребенка может отставать от возрастной нормы, но в ряде случаев умственные способности взрослого с данным синдромом не уступают таковым у здорового человека. Часто наблюдается отсутствие молочных желез и сосков (либо их аномальные форма и расположение). Иногда ангидротическая эктодермальная дисплазия осложняется врожденной глухотой.
Диагностика
Диагностика заболевания производится на основе обследования ребенка у генетика, ДНК-исследования и изучения наследственного анамнеза. При осмотре пациента на патологию указывают характерные сочетания признаков и объективный статус больного. Генетическое определение заболевания сводится к прямому секвенированию последовательности гена EDA с целью выявления мутаций; изучение других, более редких мутаций, ассоциированных с ангидротической эктодермальной дисплазией, в настоящий момент не производится.
При изучении наследственного анамнеза особое внимание уделяют объективному статусу матери – нередко у нее, как у носительницы мутантного гена, обнаруживаются стигмы дизэмбриогенеза. К ним относят сухость кожи, ослабленные тонкие волосы, гипоплазия молочных желез, из-за чего возникают проблемы с кормлением ребенка.
Генетическая диагностика носительства мутантной формы гена EDA сопряжена с определенными сложностями, так как метод прямого секвенирования в таком случае часто дает ложноотрицательные результаты. Поэтому для этой цели используются другие методики генетического анализа – например, мультиплексная лигазная реакция.
Лечение ангидротической эктодермальной дисплазии
Специфического лечения данной патологии не существует, терапия сводится к поддержке нормальной жизнедеятельности и профилактике осложнений. Для увлажнения кожи используют специальные фармацевтические или косметологические кремы, аномалии зубов исправляют при помощи протезирования. Из-за нарушения потоотделения крайне опасным становится перегрев, поэтому особую осторожность необходимо проявлять в летние жаркие месяцы. Больных ангидротической эктодермальной дисплазией в этот период желательно содержать в кондиционированном помещении, можно оборачивать влажной простыней для увлажнения, давать обильное питье. Также проводят лечение и профилактику вторичных бактериальных и грибковых инфекций кожи, иммуномодулирующую терапию. Для профилактики глазных нарушений необходимо регулярное использование увлажняющих капель.
Прогноз
Прогноз заболевания зависит от степени выраженности сопутствующих нарушений и своевременного выявления патологии. В большинстве случаев, если диагноз был установлен в раннем возрасте ребенка и были предприняты профилактические меры (борьба с перегревом, вторичными инфекциями), то прогноз в целом благоприятный. В плане интеллектуального развития прогноз чаще всего неоднозначный – с одинаковой вероятностью возможна как умственная отсталость (олигофрения), так и сохранение когнитивного развития.
Публикации в СМИ
Дисплазия эктодермальная
Эктодермальные дисплазии — врождённые дефекты структур эктодермального генеза (в т.ч. кожи и её придатков) — наблюдают в виде нескольких самостоятельных форм и при ряде заболеваний, различающихся по клинической картине. Факторы риска • Перегрев организма • Нарушение питьевого режима.
Основные формы
• Криста–Сименса–Турена синдром (*305100, Xq13.1, дефект гена EDA, À рецессивное): врождённое сочетание отсутствия потовых желёз, частичного или полного отсутствия зубов, гипотрихоза, деформации костей носа, хейлита и синеватой пигментации кожи. У женщин-носительниц при йодной пробе характерно распределение потовых желёз в форме спиралей или V-образно, часто более выраженное на одной стороне тела. Синонимы: эктодермальная дисплазия гипогидротическая, Криста–Сименса синдром, дисплазия эктодермальная ангидротическая, Сименса синдром, кератоз идиопатический многоформный.
• Дисплазия эктодермальная гидротическая (*129500, 13q11–q12.1, дефект гена HED, Â ): аномалия развития эктодермы, проявляющаяся дисплазией эпидермиса и придатков кожи — дистрофия зубов, рахит, хейлит, конъюнктивит, врождённая дистрофия ногтей и волос (утолщение ногтей, урежение или отсутствие волос на голове), часто сопровождается кератодермией ладоней и стоп, гиперпигментацией кожи, косоглазием и умственной отсталостью; потоотделение не нарушено. Синонимы: синдром Клустона, Франческетти эктодермальная дисплазия, Клустона гидротическая эктодермальная дисплазия. Примечание. Возможна ассоциация синдрома Клустона с глухотой вследствие протяжённой делеции, захватывающей области гена HED и гена коннексина (121011, 13q12).
Специальные исследования • Рентгенография челюстей (при подозрении на отсутствие зачатков зубов) • Пробы на потоотделение (крахмально-йодная проба Минора, проба с электрофорезом пилокарпина) • Исследование волос под микроскопом: характерный ланугоподобный стержень волоса без коркового слоя • Запрещается проведение тепловой стимуляции потоотделения и пробы на терморегуляцию у детей до года.
Дифференциальная диагностика • Эллиса–ван Кревельда синдром • Лангера–Гедиона синдром.
Лечение хирургическое • Челюстно-лицевая хирургическая коррекция • Ортопедическое лечение при пороках развития конечностей.
МКБ-10 • Q77.6 Хондроэктодермальная дисплазия • Q82.4 Эктодермальная дисплазия (ангидротическая).
Код вставки на сайт
Дисплазия эктодермальная
Эктодермальные дисплазии — врождённые дефекты структур эктодермального генеза (в т.ч. кожи и её придатков) — наблюдают в виде нескольких самостоятельных форм и при ряде заболеваний, различающихся по клинической картине. Факторы риска • Перегрев организма • Нарушение питьевого режима.
Основные формы
• Криста–Сименса–Турена синдром (*305100, Xq13.1, дефект гена EDA, À рецессивное): врождённое сочетание отсутствия потовых желёз, частичного или полного отсутствия зубов, гипотрихоза, деформации костей носа, хейлита и синеватой пигментации кожи. У женщин-носительниц при йодной пробе характерно распределение потовых желёз в форме спиралей или V-образно, часто более выраженное на одной стороне тела. Синонимы: эктодермальная дисплазия гипогидротическая, Криста–Сименса синдром, дисплазия эктодермальная ангидротическая, Сименса синдром, кератоз идиопатический многоформный.
• Дисплазия эктодермальная гидротическая (*129500, 13q11–q12.1, дефект гена HED, Â ): аномалия развития эктодермы, проявляющаяся дисплазией эпидермиса и придатков кожи — дистрофия зубов, рахит, хейлит, конъюнктивит, врождённая дистрофия ногтей и волос (утолщение ногтей, урежение или отсутствие волос на голове), часто сопровождается кератодермией ладоней и стоп, гиперпигментацией кожи, косоглазием и умственной отсталостью; потоотделение не нарушено. Синонимы: синдром Клустона, Франческетти эктодермальная дисплазия, Клустона гидротическая эктодермальная дисплазия. Примечание. Возможна ассоциация синдрома Клустона с глухотой вследствие протяжённой делеции, захватывающей области гена HED и гена коннексина (121011, 13q12).
Специальные исследования • Рентгенография челюстей (при подозрении на отсутствие зачатков зубов) • Пробы на потоотделение (крахмально-йодная проба Минора, проба с электрофорезом пилокарпина) • Исследование волос под микроскопом: характерный ланугоподобный стержень волоса без коркового слоя • Запрещается проведение тепловой стимуляции потоотделения и пробы на терморегуляцию у детей до года.
Дифференциальная диагностика • Эллиса–ван Кревельда синдром • Лангера–Гедиона синдром.
Лечение хирургическое • Челюстно-лицевая хирургическая коррекция • Ортопедическое лечение при пороках развития конечностей.
МКБ-10 • Q77.6 Хондроэктодермальная дисплазия • Q82.4 Эктодермальная дисплазия (ангидротическая).
Эктодермальная гидротическая дисплазия
Диагностика зачастую заключается в клиническом наблюдении, а также исследовании генеалогического дерева с определение типа передачи: аутосомно-доминантного или рецессивного. Эктодермальная дисплазия может возникать у любых рас, но наиболее часто встречаются у европеоидов.
С клинической точки зрения могут быть выделены две основные формы:
1. Гипогидротическая форма
2. Гидротическая форма
Гипогидротическая эктодермальная дисплазия является одним из самых распространенных наследственных заболеваний и примерно встречается с частотой 1:17000 по всему миру. Проявляется синдром типичной триадой: гипогидроз, гипотрихоз и гиподонтия. Обычно патология наследуется сцеплено с Х хромосомой. У мужчин заболевание проявляется тяжелее, чем у женщин. В гидротической форме экдодермальной дисплазии поражаются обычно зубы, волосы и ногти, в то время как потовые железы проявляют сверхразвитие. Обычно такая форма наследуется аутосомно-доминантно. DJB16, регулирующий синтез белка 6 (conexin-30), является единственным известным геном, ассоциированным с гидротической эктодермальной дисплазией. У большинства людей, страдающих от синдрома, один из родителей так же является больным. Потомство заболевшего имеет 50% вероятность наследования мутантного гена. Имеется данным и об аутосомно-рецессивном типе наследования.
Основные «лицевые» черты эктодермальной дисплазии следующие: крупная лобная кость, сплюснутый нос, выраженные надглазничные валики, несимметрично посаженые уши, вдавленная средняя часть лица, нижняя треть лица уменьшена из-за недостаточного развития альвеолярной кости, губы выпуклые.
Цефалометрические исследования Vierucci и коллег показали значительную разницу в особенностях черепа больных и здоровых детей. В ротовой полости наиболее явным признаком является олигодонтия, отсутствие 6 и более зубов включая 3-и моляры. Зубы в переднем сегменте нижней и верхней челюстей конические или заостренные по форме. Эмаль также может быть дефектной. Наблюдается широкая срединная диастема и гипоплазия уздечки губы.
Обычно в заднем сегменте обнаруживается только один моляр с бочковидной коронкой. Косметическое лечение показано практически всем пациентам, а изготовление протезов уже требуется с 2-х летнего возраста пациента. В течение всего периода взросления необходима частая смена протезов, а затем постановка имплантатов, как только челюсть полностью сформируется. На настоящий день методика удаления имеющихся зубов с заменой на имплантаты является достаточно широко распространенной. Как альтернативный вариант возможно покрытие имеющихся зубов коронками в совокупности с ортодонтическим лечением. По причине сложности таких случаев рекомендуется привлечение разнопрофильных специалистов. Данный клинический случай описывает реабилитацию пациента с эктодермальной дисплазией, тяжелой атрофией костной ткани и пневматизацией верхнечелюстной пазухи при помощи имплантатов.
Описание клинического случая
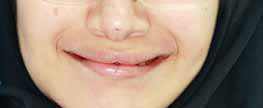
В клинику обратилась 21-летняя пациентка с генетически обусловленной эктодермальной дисплазией для лечения частичного отсутствия зубов на верхней и нижней челюсти при помощи установки имплантатов. Исследование генеалогического древа выявило такое же заболевание у бабушки, отца, дяди и брата пациентки (Фото 1). Основные жалобы: нарушение эстетики и затрудненное пережевывание пищи. В анамнезе имеется анемия, других патологических состояний, включая аллергию на медикаменты, не выявлено. Также девушка сообщила о приеме фолиевой кислоты под надзором терапевта. В истории болезни отмечено нормальное потоотделение с рождения и нормальная толерантность к теплу (Фото 2).
Фото 1. Генеалогическое древо
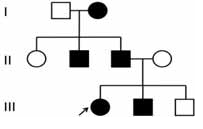
Фото 2. Фронтальный вид
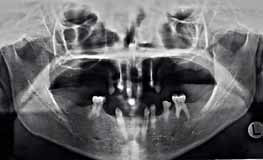
Клинический осмотр выявил редкие и мягкие волосы на голове, медленные рост ногтей, множественное отсутствие зубов (без учета третьих моляров, по классификации ADA): 2, 3, 4, 5, 12, 13, 14, 15, 18, 23, 24, 25, 26, 28, 29 и 31. Гипоплазия эмали обнаружена у 13 и 23. На ОПГ выявлена аномальная структура зубов 7, 8, 9, 10 и 21. Также горизонтальная убыль кости в заднем участке нижней челюсти, возвышение зубов 19 и 30, утрата вертикальной высоты, неразвитость альвеолярного гребня и двустороннее расширение верхнечелюстной пазухи (Фото 3 и 4).
Фото 3. Внутриротовой фронтальный вид
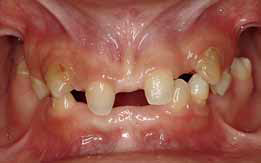
Фото 4. ОПГ до операции
— Эктодермальная дисплазия (гидротическая форма)
— Частичная олигодонтия на верхней и нижней челюстях
— Утраченная вертикальная высота
— Эстетическое нарушение, жевательная и речевая дисфункция
— Двусторонняя пневмотизация верхнечелюстных синусов
План лечения верхней челюсти включал удаление всех зубов, двусторонний синуслифтинг и наращивание альвеолярной костной ткани.
Затем изготовление временного протеза, установка имплантатов, фиксация протеза на имплантаты при помощи цемента. На нижней челюсти запланирована фиксация протезов на собственные зубы, а также имплантаты 28 и 29.
Первоначально были изготовлены предварительные слепки, фиксирована центральная окклюзия и определена протетическая плоскость. Отлиты модели, также изготовлен прикусной валик для фиксации вертикальной высоты. При помощи КТ оценен уровень кортикальной кости для потенциальной локализации имплантатов (Фото 5 и 6). В конечном итоге пациенту представлен лечебный план. После получения согласия проведено препарирование зубов нижней челюсти и постановка временного протеза. При помощи прикусного валика определена центральная окклюзия для изготовления протеза на верхнюю челюсть (Фото 7).
Фото 5: КТ скан верхней челюсти до вмешательства

Фото 6: КТ скан нижней челюсти до вмешательства
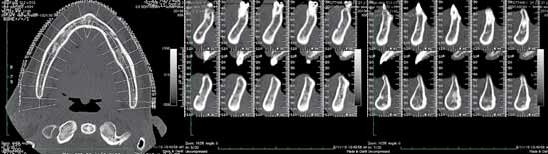
Фото 7: Временный протез и прикусной валик
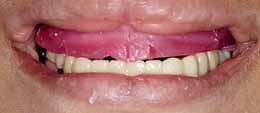
Фото 8: После установки имплантатов
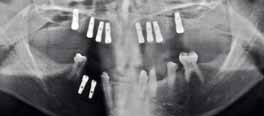
Фото 9: а) металлический каркас в артикуляторе; b и c) фиксированные на цемент протезы на верхнюю и нижнюю челюсти; d) центральное соотношение
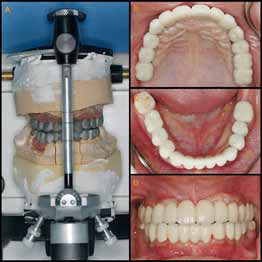
Фото 10: ОПГ после лечения
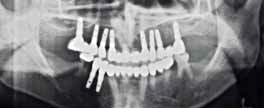
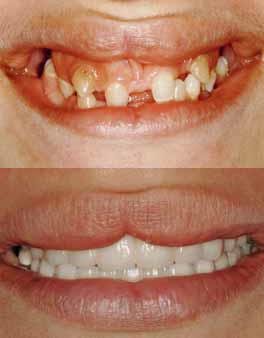
Фото 11: Фото до проведенного лечения и после
Обсуждение
Для определения синдрома эктодермальной дисплазии необходимо поражение по крайней мере двух структур: волос, зубов, ногтей или потовых желез. Однако на настоящий момент не существует единого согласия о точном числе симптомов, необходимых для постановки данного диагноза. В описанном клиническом случае диагноз был поставлен на основе поражения волос, медленного роста ногтей и олигодонтии. Патология потовых желез не установлена, обнаружен генетический аспект заболевания с аутосомно-доминантным типом наследования. Обычно лечение данных патологий состоит в изготовлении различного рода съемных и несъемных протезов. В некоторых группах пациентов также возможно использование имплантатов для фиксации протезов. Реабилитация таких пациентов весьма сложна, так как требует привлечения разнопрофильных специалистов. Также следует учитывать возраст пациента, стадию роста, наследственные особенности, дефекты мягких тканей, присутствие зубов с нарушенной формой, крупные диастемы и психологический фактор. Как правило, у этих пациентов нарушена структура альвеолярного гребня, иногда называемого «острием ножа», что, конечно же, будет затруднять процесс имплантации. Именно поэтому зачастую требуется проведение предварительной костной пластики и синуслифтинга. Существуют эстетические, функциональные и психологические аспекты, которые делают особенно важным лечение в раннем возрасте. Хотя иногда стоматологу приходится сталкиваться с особыми трудностями в виду постоянного роста пациента. Однако это не умаляет важности ортопедического лечения для благоприятного психологического состояния пациента.
Заключение
Лечение пациентов с тяжелой формой олигодонтии по причине эктодермальной дисплазии будет зависеть от уникальных анатомических особенностей, состояния зубочелюстной системы и возраста пациента. Описанный клинический случай является типичным примером реабилитации таких пациентов с привлечением разнопрофильных специалистов. Более того трехлетнее наблюдение за данным пациентом показало, что при тщательном планировании лечение с применением имплантатов является самым успешным и эффективным из всех существующих методов. Эстетическое восстановление зубочелюстной системы при синдроме эктодермальной дисплазии помогает поднять личную самооценку и в целом улучшить здоровье полости рта.
Авторы: Dr. Adel Jragh, Dr. Hassan Mousawi, Dr. Manar Al-Nouri